| Register or Login: | Password: |
|
||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||||
| ||||||||||||||
| This Document | |||||
| SummaryPlus | |||||
| Full Text + Links | |||||
| ·Full Size Images | |||||
| PDF (61 K) | |||||
| Actions | |||||
| Cited By | |||||
| Save as Citation Alert | |||||
| E-mail Article | |||||
| Export Citation | |||||
Opinion
Virulence and pathogenesis
Robin A. Weiss
Dept of Immunology and Molecular Pathology, University College London, 46 Cleveland Street, London, UK W1T 4JF
Available online 3 July 2002.
Why do viruses cause disease? As intracellular
parasites they grow at the expense of the host, yet many infections are
non-virulent. We tend to focus on unusual outcomes of infection that
are important to the individual but trivial for host–parasite
evolution, for example, paralytic polio or viral cancer. The assumption
that the features of disease help onward transmission of the virus is
true for, say, rabies, but not for AIDS or neurodegenerative diseases.
Moreover, minor host differences can result in major changes in
pathogen virulence. Although viral burden relates to disease severity,
pathogenesis is not necessarily coupled with transmission dynamics.
Why do some viruses cause disease while others have become avirulent? Here, the hypothesis that virulence usually results from natural selection acting on transmission is challenged.
Author Keywords: virulence; pathogenesis; selection; transmission dynamics; viral persistence; cross-species transmission
Subject-index terms: Virology; Microbiology; Evolution
![]()
Most of the articles in this issue are devoted to molecular pathogenesis and the host response to virus infections, in other words, how viruses cause damage to their hosts. I wish to debate why they cause damage, and to challenge the notion that virulence is usually the result of natural selection acting on transmission. Of course, these concepts are not special to virus infections; they are equally pertinent to other infectious microorganisms and parasites, but I shall restrict my discussion to viruses of vertebrates.
Virulence and pathogenesis are not quite the same thing. The former is viewed from the point of view of the virus and latter from its effect on the host [1]. Evolutionary biologists like the term virulence; molecular virologists would sooner mention pathogenesis, at least when writing a grant application or an article for TIM! Thus virulence and pathogenesis represent two sides of one coin, but why do viruses cause damage? If the answer is that they propagate at the expense of their host, why, then, do many ‘successful’ viruses not cause disease? It is probably no coincidence that two ‘harmless’ viruses of humans, TT virus (TTV) and GB virus C (GBV-C), were only discovered in the 1990s. They were revealed by molecular cloning methods while searching for non-A, -B and -C causes of hepatitis [2 and 3], but appear to be avirulent passengers.
The old adage, repeated in many medical textbooks, is that ‘well-adapted’ parasites are relatively harmless to their hosts. Although long-equilibrated viruses have in many cases become attenuated, a general assumption of evolution towards avirulence is not valid because the probability of onward transmission could be dependent on factors related to virulence, such as viral load. For example, the live, attenuated Sabin vaccine strains of poliovirus tend to revert towards neurovirulence during passage through infants, such that non-immunized contacts can acquire virulent infection. In the Americas, where wild-type virulent poliovirus has been successfully eradicated, the few cases of paralytic polio that occur each year are attributable to secondary or tertiary transmission of virulent revertants of the vaccine strain. This has led to arguments for switching to immunization with killed Salk vaccine, as still used in Scandinavian countries. The impediment to its worldwide use is the need for inoculation and booster doses.
Neo-Darwinian evolutionary biologists go further, and hold that virulence is an outcome of successful adaptation. Ebert and Hamilton [4], for example, have argued that there is a trend towards evolution of virulence in parasite–host relationships. Ewald [5] appears to think that all disease manifestations in the host directly result from selective evolution of the pathogen for efficient transmission, an over-simplistic view in my opinion. Perhaps the most valuable commentary on virulence and transmission dynamics is to be found in Anderson and May [6]. They estimate that virulence can indeed affect onward transmission of a virus or parasitic organism with a formula relating to its basic reproductive rate, R0:
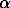 is the disease-induced host mortality or morbidity rate,
is the disease-induced host mortality or morbidity rate, 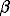 is the transmission coefficient,
is the transmission coefficient,  is the recovery rate,
is the recovery rate, 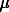 is the mortality rate for all causes, and N is the total population
size. If neither the transmission rate nor the recovery rate depend on
virulence, then R0 is maximised by making infinitesimal, that is, non-pathogenic.
is the mortality rate for all causes, and N is the total population
size. If neither the transmission rate nor the recovery rate depend on
virulence, then R0 is maximised by making infinitesimal, that is, non-pathogenic.In many cases, the particular signs and symptoms of disease can be directly related to the probability of onward transmission (Table 1). Thus, it pays a parenterally transmitted virus such as rabies to induce aggressive, rabid behaviour in its newly infected host, as well as to secret copious amounts of infectious progeny in a salivary foamite. The pathology of enteric viruses can also promote spread, as in the diarrhoea in rotavirus infection and the vomiting that follows infection by small, round Norwalk-like agents. Artificial means of transmission might also have accelerated the emergence of human viral pathogens [7].
Table 1. Signs of disease in relation to virus transmission
Alternatively, many pathogenic sequelae of infection do not relate directly to the mode of transmission of the virus (Table 1). Thus, paralytic poliomyelitis does not in itself aid transmission of the virus, although a high viral load can lead both to neurovirulence and to higher excretion of infectious virus. Moreover, signs of illness such as fever can be host responses that are useful in overcoming acute infection. The aches and fatigue during influenza are more likely to be a consequence of raised levels of interferon than of the virus itself.
Other neurovirulent virus infections are also unlikely to bear any direct relationship with transmission. Rare manifestations, such as sub-scelerosing panencephalitis following measles virus infection or progressive multifocal encephalopathy following JC papovavirus infection, are also irrelevant to transmission. I would call these examples coincident pathogenesis. Likewise, the rare occurrence of tumours decades after infection by oncogenic viruses is an oddity that benefits neither virus nor host. The oncogenic viruses come equipped with genes that induce cell proliferation or inhibit apoptosis, which aid viral replication. These could play a role in subsequent tumorigenesis in a small proportion of the infected hosts, but these late events of infection have little or no selective effect on viral or host populations.
An interesting example of the uncoupling of virulence and transmission is the H5N1 avian influenza virus discussed by Hatta and Kawaoka (this issue). This highly pathogenic variant apparently arose anew within the live bird markets of Hong Kong, possibly by a duck or quail virus adapting to chickens. Within the chicken population, H5N1 was both highly virulent and highly contagious. In humans, however, it proved almost as virulent, killing six of the 18 infected persons, yet there was no human-to-human transmission. Of course, had a human influenza virus been prevalent at the same time, a reassortant virus could have acquired the virulence genes of the avian virus with the transmissibility genes of the human virus, but that only goes to show that virulence within the infected host and transmissibility between hosts do not invariably go hand in hand.
Acute infections that do not persist in the individual host require a large reservoir host population in which to survive. Therefore host populations that were derived from small founder numbers often lacked pathogens found in major, contiguous populations of the same species. The humans who first colonized the Americas by migrating across the Bering Straits, and those who populated the Pacific islands, did not bring measles or smallpox viruses with them. Either they could not sustain such infections, or they set out for these destinations before those viruses had transferred from their former animal hosts [8 and 9]. Thus they had developed neither herd immunity nor host-selection for resistance to disease. They were decimated once these viruses were introduced, leading Darwin to comment in his Voyage of The Beagle, ‘Wherever the European has trod, death seems to pursue the aboriginal’.
Ebert and Hamilton [4], however, think that more often than not, when viruses or other microorganisms cross into new host populations or new host species, they are ill-adapted to take off, and peter out unnoticed. The great pandemics of plague, smallpox, cholera and HIV, they would argue, are the newsworthy exceptions, and there must be thousands of failed crossovers that have never come to light. Although it is not possible to enumerate the failures, I wonder whether increased pathogenesis, rather than no replication, is more the rule than the exception ( Table 2).
Table 2. Examples of changes in virulence upon cross-species transmission of virusesa
Avirulence
could be a particular feature of persistent infections that have
co-evolved for millions of years with their ‘natural’ hosts.
Cross-species transfer can then be catastrophic (Table 2).
Thus, herpesvirus B causes little worse than cold sores in macaques but
induces a lethal encephalitis in humans. The disastrous cross-species
infection of -herpesviruses between African and Indian elephants is a similar example [10].
Why are simian lentiviruses relatively non-pathogenic in their African
hosts of provenance, while they cause AIDS in new hosts such as humans
and macaques? We do not know the mechanisms, but the subtlety of immune
responses could provide some of the answers. The intimate
co-exploitation of immune modulation, discussed by Mocarski (this
issue) for cytomegalovirus and by Otten et al. (this issue) for
murine mammary tumour virus, illustrate how persistent viruses subvert
host responses to their advantage. But this subversion is only partial;
it can become completely unregulated in the immunocompromised host, or
upon cross-species infection.
When persistent infection leads to pathology, this can be a rare event or can occur beyond the natural life-span of the host, as discussed already for oncogenic viruses. Many persistent virus infections that are not latent are kept well in check by cell-mediated immunity despite their mechanisms to evade acute immune responses. Such virus infections, then, become pathogenic only in immunocompromised individuals, such as transplant recipients and AIDS patients, exemplified by many of the human herpesviruses. But for other persistent infections, the immune reactions to them could be the actual cause of pathogenesis: immunopathology is a feature of lymphocytic choriomeningitis virus infection in mice, and possibly of hepatitis B virus infection in humans.
Persistent infections can afford to be less contagious than acute infections, but eventually must be transmitted either horizontally or vertically to new hosts. Being infected does not necessarily mean being infectious, as exemplified by varicella-zoster virus. Although acute chickenpox is highly contagious among virus-naive children, recovered individuals harbour the virus in sensory ganglia for decades without being transmitters. Yet a grandparent with an episode of zoster, although much less infectious than acute chickenpox, can nonetheless pass the virus on to a new host generation and trigger another local epidemic. Persistence with occasional episodes of infectiousness is an efficient strategy for the maintenance of a virus in small or isolated host populations, such as the human hunter-gathers of times past.
Viruses can replicate to such high loads in the infected host that when this is accompanied by a high mutation rate, within-host competition and evolution become apparent [11]. The generation of defective viruses, such as the von Magnus effect with influenza virus and defective interfering particles with vesicular stomatitis virus, can limit the pathogenesis of replication-competent, wild-type genomes. High virus turnover coupled with persistence can lead to astonishingly rapid rates of genetic divergence. Thus chronic HIV-1 infection for six years in a single person yields as much variation as a year's worldwide spread of influenza among millions of hosts [12].
Competition within a host can extend to unrelated viruses. For example, coincident human infection by GBV-C and HIV appears to protect against or delay progression to AIDS [13 and 14]. There could be rich pickings here for mathematical biologists to model modulations of virulence [15].
With HIV, it is a moot point as to whether the divergence is genetic noise or evolution [16 and 17]. Many variants emerge, and some can be more pathogenic than others, but again this is not necessarily coupled with transmission. An HIV-infected person is likely to be most infectious to others either during primary viraemia, or in late-stage AIDS, the phases when virus load is high. In late-stage infection, however, HIV variants often emerge that use the CXCR4 coreceptor (X4 HIV) rather than CCR5 (R5 HIV). Although X4 viruses can occasionally infect a new host, R5 viruses are far more commonly transmitted. X4 viruses appear to be poorly transmissible viruses, and indeed to be at a selective disadvantage after transmission in immunocompetent hosts. They can themselves be regarded as opportunistic infections that only flourish in the immunocompromised host while exacerbating the disease [17].
This example of receptor change in relation to pathogenesis is not unique to HIV. Rossman et al. (this issue) discuss picornaviruses that use receptors of the immunoglobulin (Ig) superfamily (as HIV and human herpesvirus 7 also do in binding to CD4). Yet picornaviruses such as coxsackie B3 and B5 can use the Ig-like coxsackie–adenovirus receptor (CAR) to establish infection on mucosal surfaces but then adopt a different receptor, decay-accelerating factor (DAF; CD55), for internal, mesenchymal infection [18]. Coxsackie-induced myocarditis could well depend upon this switch of host receptor [19], and the cardiopathy is irrelevant to the virus’ transmission dynamics.
Adaptation of receptor usage might also occur within transgenic animals expressing both their own receptor and that of another species. I have warned of cross-species infection as a possible hazard in pigs transgenic for human DAF to overcome hyperacute rejection in xenotransplantation [20]. The picornavirus causing swine vesicular disease is remarkably close in sequence to coxsackie B5 [21] and could well be derived from humans in the recent past. A recent study, however, indicates that human Coxsackie B viruses that use DAF on human cells actually infect porcine cells via pig CAR rather than pig DAF [22]. Nonetheless, pig picornaviruses might move into humans via transgenic animals bearing human receptors [20].
As our molecular understanding of viral pathogenesis proceeds apace, there is a need to develop further the concepts of pathogenesis and virulence. There seems to be insufficient dialogue between evolutionary biologists and molecular pathologists. The former analyse sequence data and model transmission, whereas the latter investigate host–parasite interactions in terms of protein interactions.
We should also
prepare for some unpleasant surprises, not only of unexpected
cross-species infections such as HIV and variant Creutzfeldt–Jakob
disease (vCJD), but also in molecular pathogenesis. For instance,
engineering an interleukin (IL)-4 gene into the genome of ectromelia
virus (a murine poxvirus) was expected to attenuate the virus by
accelerating host immune responses. Instead it led to increased
virulence [23], a finding that will not have been overlooked in designing bioweapons.
![]()
1. N. Nathanson et al.Viral Pathogenesis, Lippincott–Raven (1997).
2. S. Hino , TTV, a new human virus with single stranded circular DNA genome. Rev. Med. Virol. 12 (2002), pp. 151–158. Abstract-Elsevier BIOBASE | Abstract-MEDLINE | Abstract-EMBASE | Full Text via CrossRef
3. J.N. Simons et al., The GB viruses. Curr. Top. Microbiol. Immunol. 242 (2000), pp. 341–375. Abstract-Elsevier BIOBASE | Abstract-EMBASE | Abstract-MEDLINE
4. D. Ebert and W.D. Hamilton , Sex against violence: the coevolution of parasitic diseases. Trends Ecol. Evol. 11 (1996), pp. 79–82. SummaryPlus | Full Text + Links | PDF (666 K)
5. P.W. Ewald Evolution of Infectious Disease, Oxford University Press (1994).
6. R.M. Anderson and R.M. May Infectious Diseases of Humans, Oxford University Press (1992).
7. E. Drucker et al., The injection century: massive unsterile injections and the emergence of human pathogens. Lancet 358 (2001), pp. 1989–1992. SummaryPlus | Full Text + Links | PDF (606 K)
8. Diamond, J. (1997) Guns, Germs and Steel. A Short History of Everybody for the Last 13, 000 Years, Jonathan Cape.
9. R.A. Weiss , The Leeuwenhoek Lecture 2001. Animal origins of human infectious disease. Philos. Trans. R. Soc. Lond. B Biol. Sci. 356 (2001), pp. 957–977. Abstract-Elsevier BIOBASE | Abstract-GEOBASE | Abstract-MEDLINE | Full Text via CrossRef
10. L.K. Richman et al., Novel endotheliotropic herpesviruses fatal for Asian and African elephants. Science 283 (1999), pp. 1171–1176. Abstract-EMBASE | Abstract-MEDLINE | Full Text via CrossRef
11. D.M. Dummy In: E. Domingo et al.Origin and Evolution of Viruses, Academic Press (1999).
12. B. Korber et al., Evolutionary and immunological implications of contemporary HIV-1 variation. Br. Med. Bull. 58 (2001), pp. 19–42. Abstract-MEDLINE | Abstract-EMBASE | Abstract-Elsevier BIOBASE
13. H.L. Tillmann et al., Infection with GB virus C and reduced mortality among HIV-infected patients. New Engl. J. Med. 345 (2001), pp. 715–724. Abstract-EMBASE | Abstract-MEDLINE | Abstract-Elsevier BIOBASE | Full Text via CrossRef
14. J. Xiang et al., Effect of coinfection with GB virus C on survival among patients with HIV infection. New Engl. J. Med. 345 (2001), pp. 707–714. Abstract-EMBASE | Abstract-MEDLINE | Abstract-Elsevier BIOBASE | Full Text via CrossRef
15. S.A. Frank , Models of parasite virulence. Q. Rev. Biol. 71 (1996), pp. 37–78. Abstract-GEOBASE | Abstract-MEDLINE | Full Text via CrossRef
16. M. Sala and S. Wain-Hobson , Are RNA viruses adapting or merely changing. J. Mol. Evol. 51 (2000), pp. 12–20. Abstract-MEDLINE | Abstract-EMBASE | Abstract-Elsevier BIOBASE
17. R.A. Weiss , Gulliver's travels in HIVland. Nature 410 (2001), pp. 963–967. Abstract-EMBASE | Abstract-Elsevier BIOBASE | Abstract-MEDLINE | Full Text via CrossRef
18. D.J. Evans and J.W. Almond , Cell receptors for picornaviruses as determinants of cell tropism and pathogenesis. Trends Microbiol. 6 (1998), pp. 198–202. SummaryPlus | Full Text + Links | PDF (140 K)
19. T.A. Martino et al., Cardiovirulent coxsackieviruses and the decay-accelerating factor (CD55) receptor. Virology 244 (1998), pp. 302–314. Abstract | Abstract + References | PDF (257 K)
20. R.A. Weiss , Transgenic pigs and virus adaptation. Nature 391 (1998), pp. 327–328. Abstract-MEDLINE | Abstract-EMBASE
21. G. Zhang et al., Molecular evolution of swine vesicular disease virus. J. Gen. Virol. 80 (1999), pp. 639–651. Abstract-MEDLINE | Abstract-EMBASE
22. O.B. Spiller et al., Coxsackie B viruses that use human DAF as a receptor infect pig cells via pig CAR and do not use pig DAF. J. Gen. Virol. 83 (2002), pp. 45–52. Abstract-MEDLINE | Abstract-Elsevier BIOBASE | Abstract-EMBASE
23. R.J. Jackson et al., Expression of mouse interleukin-4 by a recombinant ectromelia virus suppresses cytolytic lymphocyte responses and overcomes genetic resistance to mousepox. J. Virol. 75 (2001), pp. 1205–1210. Abstract-EMBASE | Abstract-MEDLINE | Abstract-Elsevier BIOBASE | Full Text via CrossRef
24. Weiss, R.A. Cross-species infection. Curr. Top. Microbiol. Immunol. (in press).
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Volume 10, Issue 7 , 1 July 2002, Pages 314-317 |
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Copyright © 2004 Elsevier B.V. All rights reserved. ScienceDirect® is a registered trademark of Elsevier B.V. |

